Motivation
The human body is comprised of a large collection of diverse cell types, each providing a specialized and context-specific function. The advent of high-throughput single-cell technologies have enabled unbiased categorization of cells from diverse developmental and diseased processes. In our work we seek to use single-cell tools to understand: i) how cells vary through time, ii) how these differences affect cellular decisions and iii) how transcription factors affect the activity of regulatory elements and, in turn, how these elements lead to functional differences in expression. Our lab seeks to address these challenges by developing biological tools to measure chromatin dynamics in single-cells. Further, we use these tools to study chromatin alterations in adult stem cells across normal, ageing and cancer tissues in effort to uncover new mechanisms of gene regulation and their contribution to disease.
Research Interests
Developing new technologies
Our group employs a mix set of skills relating to molecular biology, device engineering and microscopy.
We use these skills to leverage sequencing technologies to make high-throughput measurements that seek to
better understand gene regulation within living systems. For example, in previous work, we have developed
RNA-MaP and
Assay for Transpose Accessible Chromatin (ATAC-seq).
Notably, ATAC-seq has become an increasingly popular method to measure genome-wide chromatin accessibility, i.e. the epigenome.
Recently, we have also adapted this method to profile the epigenomes of single-cells
(scATAC-seq).
We are actively developing technological tools for improving the quality and throughput of these measurements,
as well as integrating scATAC-seq with other single-cell ‘-omic’ measurements.
Computation for inferring ‘causative’ gene networks
With the development of novel technologies comes the possibility of new discoveries and subsequently
the need for new computational tools. We develop computational solutions to support these novel technologies.
Notable examples include methods for high-content
image-analysis,
nucleosome calling
and single-cell epigenomics.
Now, we look to build new computational methods that focus on integrating ensemble and single-cell ‘-omics’ data to infer causative
gene networks. Such networks will seek to model governing cis and trans effectors of dynamic cell function.
Epigenomic regulation of cell fate decisions
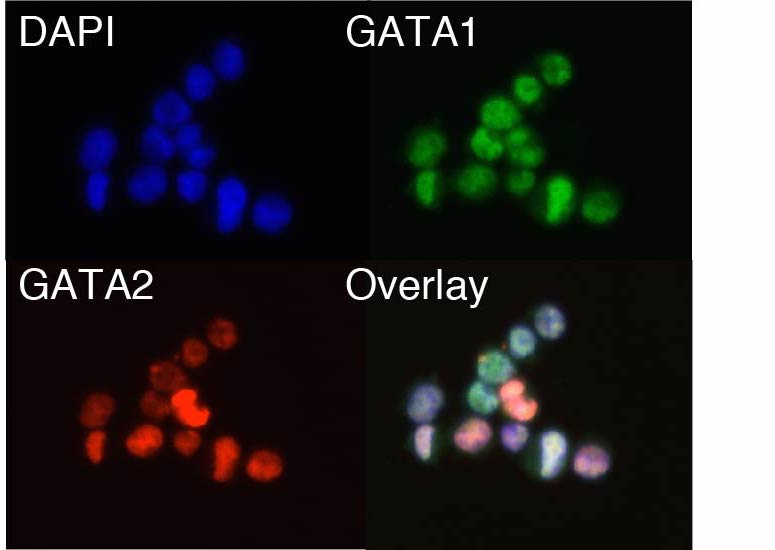
These efforts coalesce into our studies of the epigenome across cell fate decisions. In the lab we study
leukemia and early
human hematopoiesis.
However, we also collaborate broadly and we welcome new endeavors. In previous work, we have used some
of the experimental and computational methods described above to both understand the regulatory effectors that govern
hematopoietic cell fate and in addition we use these findings to better understand the ontogeny of the human cancer,
Acute Myeloid Leukemia (AML).